Example usage of PAModelpy
Example 1: setting up an Escherichia coli Protein Allocation model (PAM)
Escherichia coli (E.coli) is a commonly used model organism in Microbiology. When this microorganism is grown on increasing glucose concentration, it shifts from a purely respiratory metabolism to a respiro-fermentative metabolic phenotype. This phenomenon is called ‘overflow metabolism’. Interestingly, overflow metabolism cannot be simulated using normal genome-scale models without additional constraints. With properly parametrized protein-constrained models however, we are able to simulate this metabolic phenotype. In this example, we’ll set-up the E.coli PAM, and we’ll study the predicted metabolic phenotypes for a range of glucose uptake rates.
For this entire tutorial, you’ll need to load the following packages:
#importing the packages
import os
from cobra.io import read_sbml_model
import pandas as pd
#load PAMpy modules
from PAModelpy.EnzymeSectors import ActiveEnzymeSector, TransEnzymeSector, UnusedEnzymeSector
from PAModelpy.PAModel import PAModel
from PAModelpy.PAMValidator import PAMValidator
from PAModelpy.configuration import Config
from PAModelpy.utils import (merge_enzyme_complexes, parse_reaction2protein, _get_genes_for_proteins)
Step 1: Initiate the protein sectors
Each protein-allocation model has three sectors: active enzyme sector (enzymes catalyzing the metabolic reactions),
translational protein sectors (i.e. ribosomal proteins required for translation) and unused proteins (idle proteins
which help the cell adapt to new conditions). The total of these three sectors is limited by an upperbound. This
upperbound is determined by the sum of all non-maintenance enzymes, which is assumed to be constant for prokaryotes.
For examples on how to parametrize these sectors, refer to Scripts/create_ecolicore_pam_inclUE.ipynb.
Important note: This tutorial aims to teach you how to build a PAM from scratch, so that you can adapt the sectors where needed and you get to understand the logic of the PAModelpy package. However, in the
utilstoolbox, there is a dedicated method for building a pam with the following syntax:
def set_up_pam(pam_info_file:str = '',
model:Union[str, cobra.Model] = 'Models/iML1515.xml',
config:Config = None,
total_protein: Union[bool, float] = True,
active_enzymes: bool = True,
translational_enzymes: bool = True,
unused_enzymes: bool = True,
sensitivity:bool = True,
enzyme_db:pd.DataFrame = None,
adjust_reaction_ids:bool = False) -> PAModel
You can call this function as follows:
from PAModelpy.utils import set_up_pam
import os
pam = set_up_pam(os.path.join('Data', 'proteinAllocationModel_iML1515_EnzymaticData_py_uniprot.xlsx'),
os.path.join('Models', 'iML1515.xml'))
More information can be found in the :ref:PAM_setup_guide
1.1: Active enzyme sector
The active enzyme sector will be build using information about which enzymes catalyzes a specific reaction,
the turnover rate of the catalysis and the molar mass of the enzyme. In this example we’ll use the parameters as
published by Alter et al. (2021), which can be found in the Data folder of the PAModelpy repository
First, we’ll define the paths we’ll download the data
protein_sector_info_path = 'Data/proteinAllocationModel_iML1515_EnzymaticData_py_uniprot.xlsx'
active_enzyme_data = pd.read_excel(protein_sector_info_path, sheet_name='ActiveEnzymes'))
The data is now in a dataframe with the following columns:
rxn_id - gpr - gene - enzyme_id - molMass
We need to collect the data from this table and put it in the correct structure to be parsed into the ActiveEnzymeSector
object. The main input in this object is the rxn2protein dictionary, where all the information about protein-reaction
associations required to build the protein-reaction relations in the model. It has the following format:
{
"R1": {
"E1": {
"f": "forward kcat",
"b": "backward kcat",
"molmass": "molar mass",
"protein_reaction_relation": [["E1"]]
},
"E2": {
"f": "forward kcat",
"b": "backward kcat",
"molmass": "molar mass",
"protein_reaction_relation": [["E1"]]
}
}
}
If you have a information about the gene-protein-reaction associations (e.g. ‘AND’/’OR’ relations between different
peptides/proteins for one or more reactions), this information can be added in the protein_reaction_relation entry
of the reaction2protein dictionary. This entry is a list of lists, in which each sublist represent one functional
enzyme (complex). This means if E1 and E2 catalyze the same reaction, the protein_reaction_relation becomes [['E1','E2']]
for an enzyme complex (‘AND’ relation), and [['E1']['E2']] for isozymes (‘OR’ relation). In this example we will use
the peptide ids as defined by UniProt. The paper introducting sEnz
uses a different system, based on EC numbers. How to build those PAMs can be found in the scripts associated to the
publication and in the Script/pam_generation.py file. Now we will use gene-protein-reaction relations obtained from a
genome-scale model and uniprot to include different enzyme relations.
Fortunately, in the utils directory, there are functions available which help you parse the information to the
reaction2protein and protein2gene dictionaries. This also includes a gapfilling step which assigns a ‘dummy’ identifier to
each reaction which is not associated with an enzyme id.
Before we start we have to ensure each row contains one ‘catalytical unit’, e.g. a single enzyme or enzyme complex which is
able to catalyze a reaction. This means that we have to parse the dataframe. For this, we can use some tools provided in the utils directory.
#load the genome-scale information
model = read_sbml_model(os.path.join('Models', 'iML1515.xml'))
#get the mapping between the protein and genes
protein2gene, gene2protein = _get_genes_for_proteins(active_enzyme_data, model)
#parse the dataframe
active_enzyme_per_cu = merge_enzyme_complexes(active_enzyme_data, gene2protein)
reaction2protein, protein2gpr = parse_reaction2protein(active_enzyme_data, model)
#Use the mapping between reactions and proteins to generate the active enzymes sector
active_enzyme_sector = ActiveEnzymeSector(rxn2protein=rxn2protein, protein2gene=protein2gene)
1.2: Translational protein sector
The translational sector requires less parameters. We only need to define the reaction to which this section is proportional,
(for example the biomass pseudoreaction or substrate uptake rate), defined by id_list, and the slope (tps_mu) and
intercept (tps_0) of this relation.
translational_info = pd.read_excel(protein_sector_info_path, sheet_name='Translational')
id_list = [translational_info[translational_info.Parameter == 'id_list'].loc[0,'Value']]
translation_enzyme_sector = TransEnzymeSector(id_list=id_list,
tps_0=[translational_info[translational_info.Parameter == 'tps_0'].loc[1,'Value']],
tps_mu=[translational_info[translational_info.Parameter == 'tps_mu'].loc[2,'Value']],
mol_mass=[translational_info[translational_info.Parameter == 'mol_mass'].loc[3,'Value']])
1.3 Unused enzyme sector
The unused enzyme sector is defined in a very similar way as the translational protein sector. We assume that this section is absent when the microbe is growing at it’s highest growth rate. We’ll use this assumption to define the slope:
unused_protein_info = pd.read_excel(pam_info_file, sheet_name='ExcessEnzymes')
ups_0 = unused_protein_info[unused_protein_info.Parameter == 'ups_0'].loc[2,'Value']
smax = unused_protein_info[unused_protein_info.Parameter == 's_max_uptake'].loc[1,'Value']
id_list =[unused_protein_info[unused_protein_info.Parameter == 'id_list'].loc[0,'Value']]
unused_protein_sector = UnusedEnzymeSector(id_list=id_list,
ups_mu=[ups_0/smax],
ups_0=[ups_0],
mol_mass=[unused_protein_info[unused_protein_info.Parameter == 'mol_mass'].loc[3,'Value']])
Step 2: Building the model
Now we are ready to build the model! We’ll need to determine a maximal protein concentration. Following Alter et al. (2021), let’s take 0.258 mmol/gcdw/h. As a basis, we’ll use the iML1515 model, created by Monk et al. (2017).
NB: for the more advanced users who want to run the sensitivity analysis, please ensure that the sensitivity argument
is set to True. If you are not interested in the sensitivity analysis, you can set sensitivity to False, which
will speed up the computation time.
#load the genome-scale information
model = read_sbml_model(os.path.join('Models', 'iML1515.xml'))
#load the PAM with the genome-scale information and the information about the enzyme sectors
pamodel = PAModel(id_or_model=model,
p_tot=0.258,
active_sector=active_enzyme_sector,
translational_sector=translation_enzyme_sector,
unused_sector=unused_protein_sector,
sensitivity =True
)
Step 3: run and validate the model results
To see if the PAM is working we can run some dummy simulations. Also, the PAMValidator module has functions which allow for easy visualization of the model predictions vs measured experimental data.
pamodel.test()
This is a simulation with a glucose uptake rate set to 10 mmol/gcdw/h. We can easily change to a different substrate uptake rate, e.g. 5 mmol/gcdw/h by putting that in as an function argument
pamodel.test(5)
In the PAMValidator object, you can find functions to run simulations over a range of substrate uptake rates.
To initiate the PAMValidator, you need to provide the model and a path to an excel file with experimental data.
In this example,we’ll use the experimental data which can be found in
Data/Ecoli_phenotypes/Ecoli_phenotypes_py_rev.xls.
from PAModelpy.PAMValidator import PAMValidator
validator = PAMValidator(pamodel,'Data/Ecoli_phenotypes/Ecoli_phenotypes_py_rev.xls')
#model flux rates of biomass formation, acetate, CO2 and O2 vs glucose uptake rate for a range of growth rates
validator.validate_range()
Alternatively, you can run simulations in a good old-fashioned for-loop. For examples on how to do that look at the
jupyter notebooks in the Figures directory.
Step 4: interpreting the results
What does the result tell you? What is the predicted metabolic phenotype and how does this relate to the experimental results. Did the model capture overflow metabolism?
Outlook
After this tutorial, you know how to apply PAModelpy to this very well-studied E.coli example. But how to address you’re
specific issue with your specific microbe? In the next example we’ll show you how to use the Config() object to give the
PAModel the right naming conventions for your specific microbe. Are you more interested in performing modifications to
the model, such as deleting or adding enzymes, changing kcats, changing enzymes upper- and lowerbounds? Then have a look
at the following jupyter notebook: Examples/PAModel_example_script.ipynb. Have fun!
Example 2: working with the Escherichia coli Protein Allocation model (PAM)
In Example 1, a detailed step-by-step guide for building a PAM is given. In this example we use methods available in PAModelpy to build a PAM and give you an overview of things you can do with the PAM once it is build.
1. Building the PAM using PAModelpy.utils
In the example above, you’ve learned that there are quite some steps required to build a PAM. However, in PAModelpy, there are methods available which help you do this. For these methods, you need a specific way of structuring the information about the enzymes sectors. This makes it easier to automate generation of PAMs. The details are discussed in the PAM_setup_guide. For this example, we use the methods describes in the PAM_setup_guide to build a pam.
from PAModelpy.utils import set_up_pam
# Define input paths
model_path = "Models/iML1515.xml"
param_file = "Data/proteinAllocationModel_iML1515_EnzymaticData_new.xlsx"
# Build the PAM
pam = set_up_pam(pam_info_file=param_file,
model=model_path,
total_protein=0.258, # Optional: Total protein concentration (g_prot/g_cdw)
active_enzymes=True, # Do you want to include an active enzyme sector?
translational_enzymes=True, # Do you want to include a translational protein sector?
unused_enzymes=True, # Do you want to include an unused enzyme sector?
sensitivity=False, # Do you want to perform a sensitivity analysis?
adjust_reaction_ids=False) # Does your model ignore suffixes like 'pp' and 'ex'? (ofter the case in core models)
2. Modifying the pam
Here are some example modifications to the PAM
Changing kcat values
For changing a kcat value, you need to provide the enzyme-reaction-direction mapping. For example, you want to change
the kcat relating the forward reaction of 13PPDH2 with the enzyme abundance of Q46856:
rxn2kcat = {'13PPDH2':{'f':10}}
pam.change_kcat_values(enzyme_id = 'Q46856',
kcats = rxn2kcat)
NB: ensure your kcat values are in the right units (1/s). The change_kcat_value function will convert the kcat values to the model units for you.
Changing enzyme concentrations
enzyme = pam.enzymes.get_by_id('Q46856')
enzyme.concentration = 0.1 #mmol/gCDW
Changing the enzyme sector linear equations
If you want to do modifications to the linear relations of a sector, you can use the buiild-in function to do this. This is for example usefull when you are changing carbon source and want to modify the translational sector. The units of the intercept should be in g_protein/g_CDW. The units of the slope*lin_rxn should yield g_protein/g_CDW.
translation_sector = pam.sectors.get_by_id('TranslationalProteinSector')
pam. change_sector_parameters(sector = translation_sector,
slope = 0.1, #in this case: g_p*h/(g_cdw*mmol_glc)
intercept=0.1, # g_p/g_cdw
lin_rxn_id='EX_glc__D_e', #optional: change the reaction to which the sector is related
print_change = True #do you want to see the change? False by default
)
The output will look like:
Changing the slope and intercept of the TranslationalProteinSector
Changing slope from <old slope> to 10 mg/gcdw/h"
Changing intercept from <old intercepts> to 10 mg/gcdw
Changing the total protein content
You can change the total protein content available for the enzyme sectors (in grams_protein/g_CDW)
pam.change_total_protein_constraint(p_tot=0.3)
Changing reaction bounds
In order to calculate sensitivities, the mathematical structure with which the reaction bounds are defines are altered. It is therefore recommendable to use the build-in functions to change reaction bounds. Besides being more robust, this is also more straightforward. The units are similar to the model units (mmol/gCDW/h).
pam.change_reaction_bounds(rxn_id='13PPDH2',
lower_bound= 0.1,
upper_bound=0.11)
Toggling sensitivity
For some applications you need a sensitivity analysis, for some you don’t. As the sensitivity analysis can slow the computation down, it is possible to toggle the sensitivity on and off between simulations:
pam.sensitivity = True #sensitivity is on
3. Perfoming simulations with the PAM
If you are happy with the PAM, you off course want to perform simulations! With the change_reaction_bounds function
you can change the bounds of for example the biomass production reaction or the substrate uptake rate. Changing
the objective can be done in the same way as you are used to with a cobrapy model.
The only thing you have to do is optimize!
solution = pam.optimize()
4. Getting the results of the simulations
The solution which is returned by the optimize() function is the same solution object as returned by a cobrapy model.
This solution object only includes the metabolic reaction fluxes and the objective value, and NOT the enzyme concentration
and sensitivities. To access the enzyme concentrations you can use the attribute enzyme.concentration.
If you enabled the sensitivity analysis, you can access the capacity sensitivity coefficients and enzyme sensitivity coefficients after solving the model.
csc = pam.capacity_sensitivity_coefficients #pd.DataFrame with columns: ["rxn_id", "enzyme_id", "constraint", "coefficient"]
esc = pam.enzyme_sensitivity_coefficients #pd.DataFrame with columns: ["rxn_id", "enzyme_id", "coefficient"]
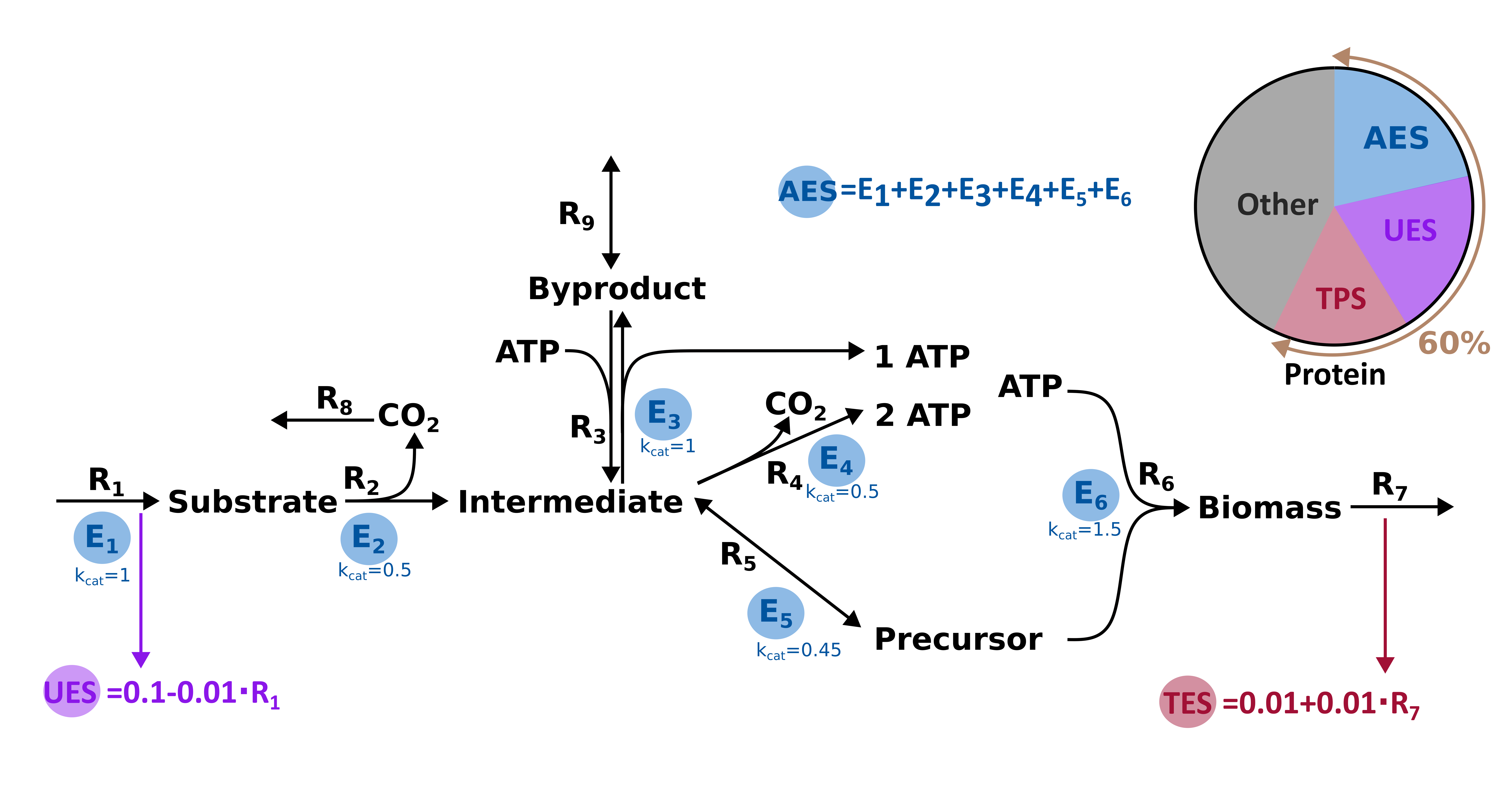
Example 3: Determining the most sensitive enzymes in a toy model
When looking at the flux distribution resulting from our simulations, we do not get any information about which enzymes played an important role in prediciting the specific metabolic phenotype. However, with the right model configurations, we get the sensitivity of the objective function to slight changes in the enzyme availability (enzyme sensitivity coefficients, ESC) as a result from the model simulations. In this example we’ll use a toy model to illustrate how these sensitivities can help us explain concepts of protein allocation.
First, all import statements you’ll need in this example:
import numpy as np
from cobra.io import load_json_model
import plotly.express
from PAModelpy.EnzymeSectors import ActiveEnzymeSector, TransEnzymeSector, UnusedEnzymeSector
from PAModelpy.PAModel import PAModel
from PAModelpy.PAMValidator import PAMValidator
from PAModelpy.configuration import Config
Step 1: Build the toy model
Obviously, we first have to build the toy model. To make it easy, we have provided the toy model structure
in a .json file in the Models directory. As the PAModelpy package makes working with real-life data easy,
it performs units conversions to some inputs. For example, the kcat value is normally published in per sec, while we need
per hour in our calculations. Furthermore, some inputs are scaled in order to decrease the order of magnitude difference
between the variables. When we want to use ‘dummy’ data in a toy model, we need to take this into account.
But before we start building the model, we need to be aware of one thing: the PAModel object assumes you want to analyse the E.coli iML1515 model by default. How can we make the model aware that we are using another model, and that we thus need other identifiers for substrate uptake rate, growth rate, etc? The Config object helps you with just that! You can use this object to configure all the identifiers you need. Don’t forget to pass this object to all the PAModel objects you’ll initialize, so all the information is passed on!
config = Config()
config.BIOMASS_REACTION = 'R7'
config.GLUCOSE_EXCHANGE_RXNID = 'R1'
config.CO2_EXHANGE_RXNID = 'R8'
config.ACETATE_EXCRETION_RXNID = 'R9'
With these defaults defined, we can start building our model.
nmbr_reactions = 9
# Building Active Enzyme Sector
kcat_fwd = [1, 0.5, 1, 1, 0.5 ,0.45, 1.5]
kcat_rev = [kcat for kcat in kcat_fwd]
rxn2kcat = {}
for i in range(nmbr_reactions-3): # all reactions have an enzyme, except excretion reactions
rxn_id = f'R{i+1}'
# 1e-6 to correct for the unit transformation in the model (meant to make the calculations preciser for different unit dimensions)
#dummy molmass
rxn2kcat = {**rxn2kcat, **{rxn_id: {f'E{i+1}':{'f': kcat_fwd[i]/(3600*1e-6), 'b': kcat_rev[i]/(3600*1e-6),
'molmass': 1e6,
'protein_reaction_relation': [[f'E{i+1}']]}
}}}
active_enzyme = ActiveEnzymeSector(rxn2protein = rxn2kcat, configuration=config)
# Building Tranlational Protein Sector
translation_enzyme = TransEnzymeSector(id_list = ['R7'], tps_mu=[0.01*1e-3], tps_0=[0.01*1e-3], mol_mass= [1], configuration=config)
# Building Unused Enzyme Sector
unused_enzyme = UnusedEnzymeSector(id_list = ['R1'], ups_mu=[-0.01*1e-3], ups_0=[0.1*1e-3], mol_mass= [1], configuration=config)
# Building the toy_pam
model = load_json_model('Models/toy_model.json')
toy_pam = PAModel(model, name='toy model MCA with enzyme constraints',
active_sector=active_enzyme,
translational_sector = translation_enzyme,
unused_sector = unused_enzyme,
p_tot=0.6*1e-3, configuration=config)
Step 2: Perform the model simulations
With the model in place, we can start our analysis. Since we are interested in which enzymes are important in different metabolic phenotypes, we want to run simulations over a range of growth rates. After each simulation we need to retrieve and store the enzyme sensitivity coefficients, so we can study them. We also will save the capacity sensitivity coefficients, which will give us information about which factor is limiting metabolism (substrate or enzyme availability). We directly save all the information we need later for plotting.
substrate_axis = list()
Ccsc = list()
Cesc = list()
x_axis_csc = list()
mu_list = list()
for substrate in list(np.arange(1e-3, 1e-1, 1e-2)):
toy_pam.change_reaction_bounds(rxn_id='R1',
lower_bound=0, upper_bound=substrate)
toy_pam.optimize()
if toy_pam.solver.status == 'optimal' and toy_pam.objective.value>0:
print('Running simulations with ', substrate, 'mmol/g_cdw/h of substrate going into the system')
substrate_axis += [substrate]
mu_list += [toy_pam.objective.value]
Ccsc_new = list()
for csc in ['flux_ub', 'flux_lb', 'enzyme_max', 'enzyme_min', 'proteome', 'sector']:
Ccsc_new += toy_pam.capacity_sensitivity_coefficients[toy_pam.capacity_sensitivity_coefficients['constraint'] == csc].coefficient.to_list()
Ccsc += [Ccsc_new]
Cesc += [toy_pam.enzyme_sensitivity_coefficients.coefficient.to_list()]
print('Sum of capacity sensitivity coefficients: \t \t \t \t \t \t \t ', round(sum(Ccsc_new),6))
print('Sum of variable sensitivity coefficients: \t \t \t \t \t \t \t ', round(sum(Cesc[-1]), 6), '\n')
for csc in ['flux_ub', 'flux_lb', 'enzyme_max', 'enzyme_min', 'proteome', 'sector']:
if csc == 'flux_ub' or csc == 'flux_lb':
x_axis_csc += [rid +'_' + csc for rid in toy_pam.capacity_sensitivity_coefficients[toy_pam.capacity_sensitivity_coefficients['constraint'] == csc].rxn_id.to_list()]
else:
x_axis_csc += [rid +'_' + csc for rid in toy_pam.capacity_sensitivity_coefficients[toy_pam.capacity_sensitivity_coefficients['constraint'] == csc].enzyme_id.to_list()]
x_axis_esc = toy_pam.enzyme_sensitivity_coefficients.enzyme_id.to_list()
Step 3: Plot the enzyme and capacity sensitivty coefficients heatmaps
By plotting our results, we learn which individual reactions and enzymes contribute the most to which metabolic phenotype.
def print_heatmap(xaxis, matrix, yaxis = None):
if yaxis is None:
yaxis = list()
for i in range(1, n + 1):
yaxis += [f'R{i}']
fig = plotly.express.imshow(matrix, aspect="auto",
x = xaxis, y = yaxis,
labels = dict(x = 'sensitivity coefficients', y='substrate uptake'))
fig.show()
print_heatmap(x_axis_csc, Ccsc, yaxis=substrate_axis)
print_heatmap(x_axis_esc, Cesc, yaxis=substrate_axis)
Step 4: Interpret the results
Compare the toy model network structure :ref:toy_model_image with the results from the heatmap. Did you expect these results? Do they make
sense? Which mechanisms to explain these observations. If the observations are not inline with you’re expectations,
you can use the enzyme sensitivities to point to the enzymatic parameters which might need to be adjusted (in this dummy
example this makes no sense off course, but in reality this is a very plausible outcome).
Outlook
This tastes like more? In our publication we use the sensitivity analysis to explain metabolic phenotypes and to pinpoint
genetic engineering examples. In the Figures folder you can find the code we used to generate these results.